Pathophysiology of Turner Syndrome
1. Introduction to Turner Syndrome
Although ovarian dysgenesis, manifesting clinically as 79% Turner patients with streak gonads will undergo spontaneous enlargement triggered by puberty. This transition is explained by the “burned-out” hypothesis. According to this hypothesis, during the early 8-20 weeks of gestation, when the gonad has the potential to differentiate into a testis in response to the criticality of the Y chromosome, the X0 germs undergo meiosis onset, which is followed by progressive depletion of oocytes. Subsequently, the ovary fails to produce essential growth factors, such as insulin-like growth factor, transforming growth factor α and β/activin A, which are essential for sustaining stroma development and oocyte apposition to the advancing ovarian surface from 10 to 20 weeks of gestation. While prenatal oocyte loss occurs mostly through apoptosis, apoptosis is markedly decreased after week 13. The number of oocytes, therefore, then declines from 5.5 × 106 to approximately 0.5 × 106 at birth.
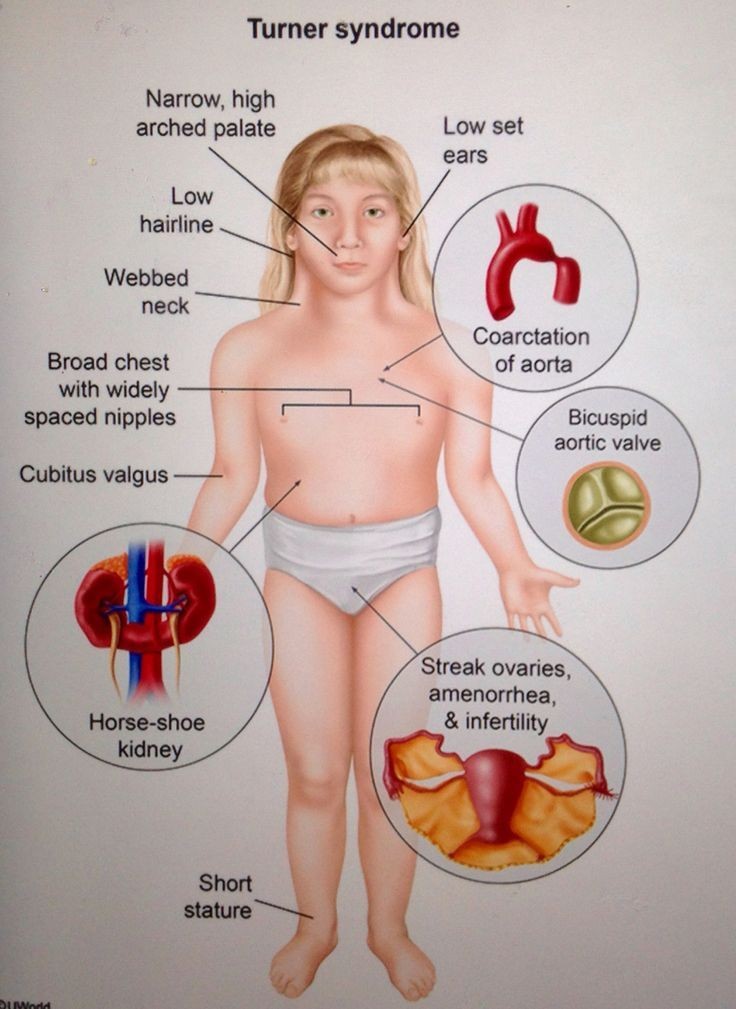
Turner syndrome (TS) is a relatively common disorder of females characterized by the absence of a whole (45,X) or part of one (45,X/46,X) of one of its two sex chromosomes (2n=46). TS is an enormous public health problem with its incidence ranging between 25-52 per 100,000 live born and 1 per 1,500 pregnancies. 60-75% of conceptions with 45,X karyotype are spontaneously aborted. The prevalence of TS in live born females can be as high as 1:200 when 45,X/46,X resolution cells. With the advent of prenatal or newborn genetic diagnosis of TS, many cases could be detected in the prepubertal period and followed up closely for potential endocrine and other disorders associated with the condition, such as short stature, gonadal dysgenesis, failure of puberty and infertility, increased risk for various vascular diseases, diabetes and obesity. In addition, non-endocrine problems that occur with increased frequency in TS include sensorineural hearing loss, autoimmunity, and intellectual disability. Therefore, early diagnosis and initiation of medical and educational intervention in children with TS is of paramount importance.
2. Genetic Abnormalities in Turner Syndrome
The haploinsufficiency of the X chromosome is the basic pathophysiology in Turner syndrome. The phenotype is mosaicism, accounting for about 60%, structural X-chromosomal anomalies for about 40%, and Y karyotype material for about 1%. It is to be realized whether the resulting clinical phenotypes are consequences of having only one single X-chromosome, or interplay of other X-linked genes due to the escape from inactivation, or mosaicism/background? Maturity of the phenotype is associated with the relative loss of tissue-specific parental origin active X. Rather than the deletion of a particular gene, the changes in the X-inactivation specific transcript (XIST) gene, Xist, and fragile X mental retardation (FMR1) gene have been considered related to Turner syndrome. Both of these are considered as the X-inactivation center manifesting the X-chromosome inactivation. Mosaicism is considered the major determinant of the different diverse presentation in Turner syndrome as a result of having two or more populations of cells as compared to a single population due to the X-deletions. It makes methylation of imprinted X-inactivation center and, in consequence, the occurrence of escape e.g., in brain tissue, which possibly is responsible for the occurrence of neurological abnormalities in Turner syndrome. These sporadic reports are therefore the signs of the heterogeneity in the etiology of those malformations present in various patients. As genetic drift would further play a role in Turner syndrome, the cases should be considered separately in the discussions, since they reflect the mosaicism and not the influence of particular genes on the phenotype. Even the recurrent deletions are not presenting the certainty, since still the mosaicism is the possible cause for further phenotypic alterations. Even among members of the same family, one can find two patients presenting differences within their phenotype. While most of the patients with a deletion of genetic material involving the SHOX gene appear as isolated cases, particularly the identical twins, their variable phenotypes are merely biased to familial cases. These suggestions highlight the fact that the quality of an X-chromosome itself varies from patient to patient and there is no precise correlation between the deleted locus located within X and severity of a phenotype. Some anomalies might also result from the micro-regions requiring the parental haploid dose. Our final assumption is that the genetic drift continuously plays a crucial role in shaping the Turner syndrome organism phenotype. Understanding Turner syndrome composition will be in the future available superimposed upon genetic alterations due to the detection of the richness of micro-RNAs, mini-satellite sequences, segmental duplications, or the “missing heritability”. Heuristic method: Try to recognize by comparing the case versus control persons the larger and smaller impacts on the phenotype. Some of the results of the thesis will be available from December 2023 onwards. In combination with the familial studies, one can compare the effects of the single fibroblastoid X chromosome with its maternal and paternal background. Up to date such mosaic cultures were interpreted in our laboratory in the framework of the dominance, which is overlapping in importance to the effects caused by the mosaicism. Often we encountered in those previous presentations the traits showing background dependent prevalence, unknown persons contributed the explanations for the diverse phenotypes to our papers or we ourselves looked for just the existing phenotypes in the publications. Up to now, we interpreted such data in the following way: parity genes, a posteriori, determine phenotypic dominance of one X; 1-4 = 2 Xm; XDict = SHOX, TCF7 abundance; additional autosomal effects. If a model proves wrong, be happy, since one is entering higher dimensional intensity with a better understanding of the nature.
3. Hormonal Imbalances and Effects
Patients with Turner karyotype 45,X present clear cytogenetic mosaicism, some of them with 46,X, i(Xq) karyotypes in 47% of the white cells, and others, 46XX in 5% of the cells. Patients with high percentages of 45,X karyotypes have the typical Turner phenotype, and some have argonemic ovaries. The percentage of 45,X karyotypes has an inverse correlation with height; a longitudinal prognosis has been proposed based on the available percentage of karyotype 45,X at the 1st year of life. In the conduct of patients with Turner syndrome, it is important to evaluate the functional impulses that are temporarily repetitive in bottles bearing tissue with only one single X chromosome; for example, hypothalamus-hypophysis, motor neuron, and lung cells.
Additional aspects of Turner syndrome might affect female reproductive potential. Ovarian non-functional germ cells should degenerate during early stages, with formation of small hyper-macronodine structures instead of oocytes. In the site, about 8% of patients undergo pubertal development and menarche regularly, at the average age of 15-16 years. Absence of estrogen might result in hypogenitalia and hypoplastic uterus. According to the difference between X chromosome centromeres, the few oocytes that persist might remain in functional silence; however, some patients might have, at least, oligomenorrhea and discrete endometrial proliferation. Uterine size varies considerably and some patients might develop regular menstruations and normal endometrial proliferation, supporting the fact that Turner patients might have, at least, relative fertility potential. More-than-expected pregnancies have been reported in experienced centers for Turner management; before development of assisted reproduction technologies, up to 8% of patients have conceived.
4. Cardiovascular and Renal Manifestations
Renal disease in TS is reported sporadically but small increases in kidney size and protein leakage are seen far more commonly during childhood. However, with the higher rates of hypertension in TS and the increased risk of developing diabetes or obesity in the future, renal disease may also be a significant risk. Studies have shown that girls with TS are at least 3 times as likely to have high blood pressure compared to control females. Even if the anatomic findings of coronary stenosis, aortic dilation, and maternal hypertension do not increase the risk of intracranial hemorrhage, chronic hypertension may. Given that chronic hypertension is more commonly seen in middle age and that girls with TS need to be followed for renal health during adulthood, it is recommended that screening for hypertension begin in childhood and continue life-long in a similar manner to the general population.
Heart disease is also a significant problem that affects women with TS in adolescent and adult age. Girls with TS have a higher prevalence of congenital heart disease as well as acquired heart disease such as hypertension (33%) and hyperlipidemia (>20%) than matched controls. The most common type of congenital heart disease is bicuspid aortic valve that occurs in approximately 10-25% of girls with TS. Careful examination of the heart beginning in infancy and then careful follow-up with echocardiograms in childhood and serially into the adult years is recommended. Although most patients with a bicuspid aortic valve have no symptoms of narrowing (stenosis) or leakage (regurgitation) of the heart valve, it is important for the parents and patient to be aware of potential heart problems in the future so that care can be provided early. MRI or CT of the heart should be considered if exercise ability in adolescents or adults with TS is significantly less than their peers. In addition, a highly specialized echocardiogram performed by an expert in adult or pediatric cardiology with knowledge of TS is best for the most accurate diagnosis. There are reports that girls with TS who are over 13 years of age and who have an abnormal Z-score of the aorta segment are at risk for increased dilation of the aortic root and should have serial (initially six mos. later and then annually) echocardiograms and possibly other imaging.
5. Psychological and Cognitive Aspects
The psycho-cognitive difficulty is a preponderant part of the clinical picture of individuals with Turner syndrome. The cognitive profile of these individuals is clear and has developmental changes, particularly in verbal-performance and social non-verbal communication. The cognitive profile is consistent and is present beyond the effect of non-verbal problems; generally, patients have non-verbal learning disabilities and a big discrepancy between the two aspects of the test. It is a neurocognitive profile that is expressed in the learning difficulties of verbal skills, processing-planning, motor coordination, daily care, management of interpersonal and social emotional abilities, and attention. There are also problems in auditory and visual processing pathways associated with particular difficulties in phonological processing.
Coordination between psychological and medical interventions has already been established. Since youth, girls with Turner syndrome (TS) are presented with diverse emotional, learning, and communication problems, either as part of the syndrome or as a result of the constant demand of treatments linked to physical health. The essentials of the typical TS endophenotype are behavioral hardship, learning disabilities, resistance to change, self-esteem problems, social and relationship skills, and communication hardship. They also exhibit mood and anxiety problems, disorganized activity behavior, angry and oppositional behaviors, in addition to habitual routines and rituals related to the diagnosis. The literature documents a range of particular problems in their interactions, which cover 56% of mental disorder diagnoses, associated with habits of self-harm (using weights) and disordered eating.