The Genetic Basis of Hemophilia A and B
1 Introduction
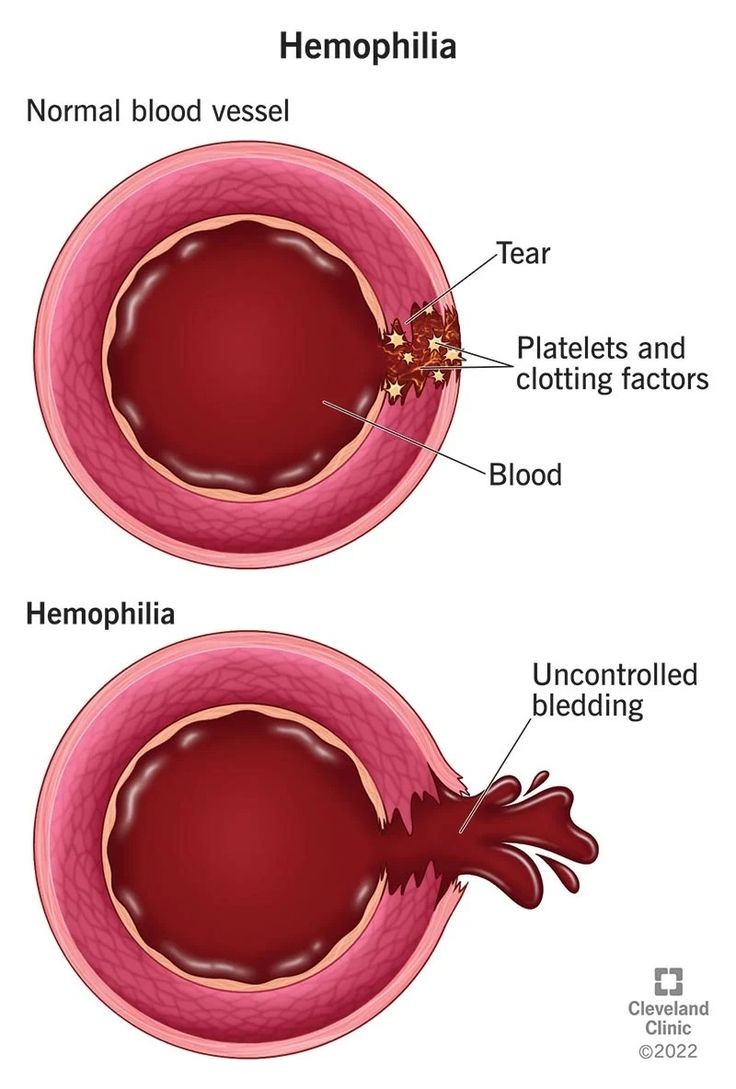
In hemostasis, the interaction of blood vessels with blood components and platelets, requiring rapid responses to vessel damage with precisely regulated control mechanisms, is essential. The complex and tightly controlled interaction among several dynamic and rapidly responding components, including blood vessels, platelets, coagulation proteins, regulators of coagulation, and excipients of the fibrinolytic system, results in thrombus formation and ensuing restitution of blood flow. Defects in the various processes that contribute to blood clot formation will be revealed as abnormal bleeding after trauma or surgery, bleeding in the mouth, epistaxis, gastrointestinal tract, muscles, soft tissues, central nervous system, or others, or in some cases, hematuria. The quantity of blood loss that is compatible with life cannot be immediately determined, and in patients with clotting abnormalities, the severity can be underestimated.
The X-linked disorders hemophilia A and B are prototypic hereditary genetic disorders with significant phenotypic and genetic variability. Affected individuals often face a lifetime of demanding medical and social challenges, including joint impairment and infectious disease. Hemostasis, physiological blood coagulation, is necessary for the maintenance of a liquid, flowing blood circulation at sites of blood vessel injury. Hemostasis involves intricate interactions among vascular, blood, humoral, and cellular components to maintain the circulation and provide the wound-healing process with the appropriate amounts of blood cells and plasma constituents. Hemorrhaging from the smallest arteriole or capillary to a larger vessel or a body cavity results from an imbalance in the processes involved.
2. Hemophilia A: Causes and Symptoms
Hemophilia A is most commonly caused by single-nucleotide mutations in the hemophilia A gene, F8, which is located on the long arm of the X chromosome at position 28, encoding the message for the factor VIII protein. In the normal human genome, F8 is 186 kb with 26 coding exons. Approximately 22% of patients with X-linked recessive inheritance have a nondeletion and noninversion defect of F8, while about 78% of patients have a definitive defect in the gene. Up to 50% of them have a family history of the disease. The condition is also characterized by a phenotypic variability in carriers, he in normal phenotypic effects, so that heterozygosity can also be dangerous and severe hemophilia do exist.
Hemophilia is a group of inherited blood disorders in which the patient has significant impairment of blood-clotting function. The condition results in spontaneous bleeds, with the most frequent sites being the joints, muscles, and internal organs. The severity of hemophilia depends on the activity level of factor VIII in the blood, and this varies from person to person. X-linked subjects with less than 1% activity levels are classified as having severe hemophilia, those with 1-5% levels are classified as having moderate disease, and those with factor VIII levels exceeding 5-40% of normal are classified as having mild hemophilia. The hallmark of the hemophilia condition is the severity and chronicity of its musculoskeletal symptoms which, even with the best care, result in a tremendous loss of physical function and the onset of musculoskeletal degeneration. Hemophilia is also known to be present in many different animal species – dogs, cats, and some breeds of horse are the most commonly affected. The vast majority of them get the disease through the same physiological pathway as humans do. This complete, naturally occurring hemophilic phenotype has been found to be beneficial for medical research and has indeed provided a unique opportunity to study this condition with a very high degree of similarity to the human disease.
3. Hemophilia B: Causes and Symptoms
In general, there are two different types of haemophilia B that have been described based on the recessive inheritance of factor IX gene mutations. The most common and severe type is haemophilia B severe. Multiple F9 gene mutations—i.e., nonsense, frameshift, and guarantee mutations—lead to premature translation of the complete or nearly complete factor IX protein. These mutations are commonly associated with the occurrence of bleeding episodes that are difficult to control. Despite the presence of a large number of bleeding events, common and/or severe spontaneous hemarthroses so that factor IX replacement therapy can be prevented. Another type of haemophilia B is mild or moderate, with a missense factor IX gene alteration effect on one or both gene alleles. Lower levels of severely decreased factor IX procoagulant activity are considered to be due to missense factor IX gene alterations. Most individuals with mild or moderate haemophilia B have minimal F9 gene alterations with hypomorphic penetrating missense cardiovascular change.
A factor IX gene alteration is usually the cause of haemophilia B. Several F9 gene alterations are known to lead to the haemophilia B phenotype. The most common cause is found at position 6 in the codon that codes for amino acid residue 148 (p.Arginine148). Changes in the 1st, 2nd, and previous 3rd positions in the p.Arg148 codon may cause a mild or severe form of haemophilia B. In affected individuals with a defect in the gene, the concentration of factor IX in the blood is very low, which is usually less than 1% of normal (and the decrease in plasma inhibitory haemostasis is less than 5%). The symptoms of haemophilia B are the same as those of haemophilia A, are triggered by trauma, and their severity depends on the level of factor IX with a deficiency and on the amount of trauma.
4. Genetic Testing and Diagnosis
For most of these techniques, the critical first step is the precise definition of the hemophilia A patient’s phenotype. Reports without precisely defined phenotype are expected to include no significant information. The use of an insufficient number of gene segments or target sequences during direct gene characterization can increase the false negative rate and decrease mutation detection performance, and may require multiple subsequent analyses which are labor and time intensive due to the large size of the F8 gene. Direct sequencing analysis of all 25 PCRs is extremely time-consuming and costly to use particularly on larger gene exons. Although incomplete DNA sequencing is known to complicate the analysis and interpretation of the results, the presence of intragenic polymorphisms can often modulate more rapidly prioritization of the top, allowing the detection of potential mutational hotspots in an increased number of candidate genes.
Several methods may be used to carry out genetic analysis in individual haemophilia A patients: Southern blotting (which is often not used today), analysis of the expression of the F8 gene, which when undetectable or reduced can be indicative of major gene rearrangements, direct analysis of the F8 gene after PCR amplification, with subsequent detection of mutations via SSCP. A preliminary screening approach detected novel disease-causing mutations with unknown effects, protein truncation test (PTT) of the abnormal phenotype (identification of DNA that corresponds to a reduced mRNA/protein level), or allele-specific oligonucleotide hybridization ASO (i.e., analysis of DNA using a PCR amplified DNA evaluation). The techniques should also be performed sequentially. The Western blotting technique is still used in limitations in the direct analysis of the F8 gene and F8 expression techniques, which, although very informative when applied, neither provides a final molecular result nor are methods either of rapid mutation detection or direct characterization, having a particularly high reversion rate.
5. Treatment Options and Future Research
Current therapeutic strategies focus on minimizing FVIII immunogenicity, such as early diagnosis and prophylactic replacement therapy. In addition, the immune response to FVIII in hemophilia A patients is driven towards the generation of inhibitory antibodies. Various strategies are currently being developed to replace FVIII therapeutics. A non-immunogenic FVIII Fc fusion protein, selected FVIII variants, small FVIII-antigenic peptide epitope IXTEN, and a method called tolerance induction by CRISPR-Cas9-based gene editing that targets Treg cells. Moreover, new techniques and products for FVIII production are being developed. Successful establishment of personalized FVIII replacement therapy will be promising. In order to develop a more efficient future treatment for hemophilia A, further studies on immune tolerance strategies need to be accomplished.
The standard treatment for patients with hemophilia A has been replacement therapy using recombinant or plasma-derived FVIII. Ideally, the measured FVIII levels should be kept higher than 1%, and the patient needs a prophylactic regime that includes the infusion of FVIII two to three times per week. Long-acting recombinant or fusion proteins can also be administered as prophylactic treatment. Recombinant FVIII is used until the appearance of inhibitory antibodies that neutralize FVIII activity and cause clinical resistance to this protein. Inhibitor formation is one of the most serious complications of current treatment and occurs in 20% to 30% of patients with severe hemophilia A during their first 50 exposure days. The presence of an inhibitor in AHA patients necessitates the use of expensive bypass agents, such as activated prothrombin complex concentrate and recombinant-activated factor VIIa, for bleeding treatment. Thus, the prevention and treatment of inhibitor formation in hemophilia A patients is an urgent issue for clinicians and researchers.