The impact of Duchenne Muscular Dystrophy on muscle cells.
1. Introduction
The role Dystrophin plays in preserving the mechanical integrity of contracting muscle has led to investigations that have typically focused on mechanical stress. However, more recently a number of non-mechanical factors have been found to directly affect the function of Dystrophin and have been shown to also lead to muscle damage, adding a new level of complexity to our understanding of this deadly disease. The purpose of this review is to focus on how non-mechanical aspects of DMD can lead to increased muscle damage. This review is not meant to be comprehensive and will mainly focus on recent data. Due to space limitations, some areas will not be reviewed in detail, including some excellent papers with information from the earlier years of research into this area.
Duchenne Muscular Dystrophy is a genetic disorder, predominantly in boys, that leads to muscle-specific degeneration and ultimately premature death. The result of the disease is a null mutation in the gene encoding the protein Dystrophin. This protein is expressed in muscle and serves to strengthen the linkage between the cytoskeletal actin filaments and the extracellular matrix basal lamina. The result of the gene mutation leads to a weakening of this connection, leading to substantial muscle damage that can result in reduced quality of life and early death without intervention. The exact role of dystrophin in muscle is not yet fully understood and remains the subject of much investigation. One likely role for dystrophin is to prevent extreme muscle damage but also to limit ongoing muscle damage caused by naturally occurring muscle stress during daily life such as muscle stretch during exercise.
2. Causes and Symptoms
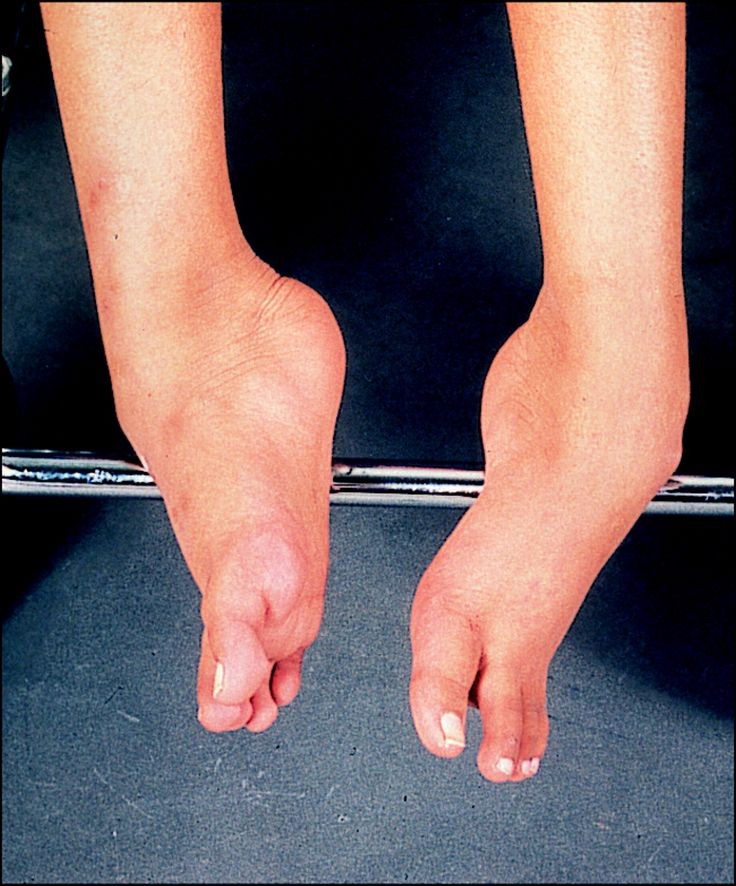
The sole muscle weakness characteristic of DMD is not the only defect that can be detected in these patients. As shown in (Fig. 1), the patients show a waddling gait, a generalized hypertrophy, calf enlargement and the lower extremities show a fibromatoid pseudohypertrophy which cannot be confused with the wide wasting and edema of calf muscles in some spinal muscular atrophies. DMD children have generalized flaccidity and laxity of joints, protruding and round heels, lumbar hyperlordosis and pseudo-Gower maneuver. As normal body movements are impossible, patients get help from anteriorly localized muscular bands of the trunk and upper extremities. The presence of a wide hypertrophy, despite severe dystrophy of the limb muscles, is one of the most confusing reasons when suspecting and screening for DMD.
Duchenne Muscular Dystrophy (DMD) is an X-linked disorder which is very common in children, with a prevalence ranging between 1:3500 and 1:5000 male births. It is an inherited pathology, and in about 1/3 of cases, there is a negative family history for DMD, due to a previous undiagnosed and unsuspected case. It has been attributed to a 1982 recessive spontaneous mutation, found in Turks, showing deletions in different parts of the gene, but also point mutations have been identified, which may produce the disease. The main clinical aspects of the pathology were described in 1868 by Guillaume Benjamin Amand Duchenne, a French neurologist, hence the name of the anomaly, who defined DMD as a sex-linked disease characterized by paralysis, excessive hypertrophy, pseudohypertrophy and initial myopathia of muscular bundles.
3. Effects on Muscle Cells
Additionally, resulting from the intervention of the immune system and the influence of stress proteins, changes in the levels and activities, the structure and the cellular localization of proteins that are involved in the maintenance and repair of the cell endoplasmic reticulum is another of the effects on muscle cells typical of DMD. These changes in the individual endoplasmic reticulum proteins correspond to an overall downregulation of expression of the endoplasmic reticulum proteins, the degree of downregulation being correlated with the severity of the DMD muscle cell phenotype. Forming, in those creatures where the problematic gene has been removed and have accelerated remodeling, that is producing less of the severe condition, less of the endoplasmic reticulum protein activity and/or faster degradation of the endoplasmic reticulum proteins, noticing a negative correlation between fibrosis and endoplasmic reticulum protein activity and extracellular matrix protein expression.
Ballistic contraction has been shown to generate a form of intracellular open trauma known as impact lesion, a well-defined lesion that then causes muscle degeneration and ultimately necrosis. This degeneration and necrosis is one of the additional effects on muscle cells typical of DMD. Apart from its inability to repair normal lesions, Duchenne muscle fails to mature and ultimately shows a large deposition of fibrous tissue. Additionally, the amount of intracellular adipose tissue increases while the capillary density decreases, as there is a decrease in vegetative fibers. In advanced myoinjury, there is loss of skeletal muscle cell membrane integrity, increased serum activities of musculoskeletal enzymes, loss of movement, and increased structural damage to skeletal muscle.
4. Current Treatments and Research
The first and the one seeing the most progress is AAV, which was the method used in the first genome edition in the liver of an adult patient with an inherited metabolic disease. In 2017 it was tested for Duchenne Muscular Dystrophy on a dog model that showed improvements to the muscle functionality of the dogs being stride length, number of steps and tail circulation in starting at less than a month after the initial treatment. Most recently, CRISPR-Cas9 has been shown to correct mutations and protein deficiency for DMD in a dish, even without the need of a correct repair template, giving translational medicine a new possibility of fast repair of genetic abnormalities. Dystrophin delivery in Duchenne’s is very complex and has been the biggest hurdle since 1988 with the discovery of the gene and the 2006 first clinical trial. Although the body mounts an immune response against re-administration of the muscle therapy, many different tested delivery methods that contained different therapeutic elements delivered as a cocktail had shown to change the course of the disease in many different DMD models and several trials have shown increases of muscle strength and the health of the heart.
Basic treatment and therapies do not address the root cause of the disease, but can help manage symptoms or ease side effects of medications. Today, a corticosteroid called prednisone is the standard of care for DMD. It works in helping with muscle strength and delay loss of function, but some children do not always respond or may not tolerate the drug. Over time most boys will lose the ability to walk independently between the ages of 10–14 and will catch up to dilated cardiomyopathy, and the children start to show fatigue and symptoms affecting the diaphragm and heart and the road to fatal. The death usually occurs, as the risk of patient choking on their vomit becomes higher over time. Life expectancy varies depending on the care a child receives, early medical care such as ventilators can lengthen how long a child lives if it becomes hard to breathe due to muscles that control breathing. Gene therapy and other repair treatments are in various stages of development both in labs and in clinical trials. Currently there are three gene delivery techniques that are being viewed as potential candidates for treatment of Duchenne.
5. Conclusion
DMD is the most common progressive and fatal form of muscular dystrophy resulting from mutations in the X-linked Dystrophin gene. Studies in the human DMD muscles have been limited due to accessibility and the scarcity of biopsy tissue, since the quadriceps muscle is often the only available motor muscle, which hinders a comprehensive analysis of the molecular mechanisms related to contractile performance in the DMD diaphragm myofibers. Determining the molecular players that contribute to DMD progression has posed a significant research effort and shed light on previously less-covered muscle groups, such as the diaphragm, to understand muscle dysfunction in DMD and perhaps create new targets for interventions before the onset of clinically significant decline in performance.
The most affected muscle group during the progression of DMD is the diaphragm, but in the later stages of the disease, muscle dysfunction frequently affects paraspinal muscles and muscles involved in respiration (scalenus, sternocleidomastoid muscles, and the intercostal muscles). Consequently, the life expectancy of individuals with DMD has posed a significant research effort to understand the consequences of dystrophic and regenerating muscle cells in the DMD diaphragm muscle. Here, we aimed to compare the gene expression profiles of human DMD versus mdx mouse, the most common animal model of DMD, as a first approach to unveil the molecular players that contribute to DMD progression, and to shed light on major differences in the expression of genes in healthy human, healthy mouse, dystrophic diaphragm, and mdx mouse.